Neurodegeneration Antibodies
Antibodies for neuro-related diseases, including Alzheimers, Parkinsons and Huntington's Disease
|
Protein aggregation in neurodegenerative disease |
Protein aggregation in neurodegenerative disease
Protein misfolding and aggregation is an intrinsic feature of many neurodegenerative diseases. From Alzheimer’s disease to rarer prion-related illnesses, protein aggregation is a basic disease hallmark that links such debilitating conditions. The proteins involved in aggregation differ in each disease (though some overlap is seen in certain cases – see table below). Moreover, it remains unclear as to whether aggregates are a cause or consequence of neurodegenerative processes. Yet, advances in the knowledge of one disease can help shed much-needed light on another. Understanding protein-folding homeostasis and how and why it goes wrong in the case of protein aggregation is invaluable to neurodegeneration research, and will hopefully provide a route to effective therapeutics targeting neurodegenerative disease.
Protein folding Homeostasis
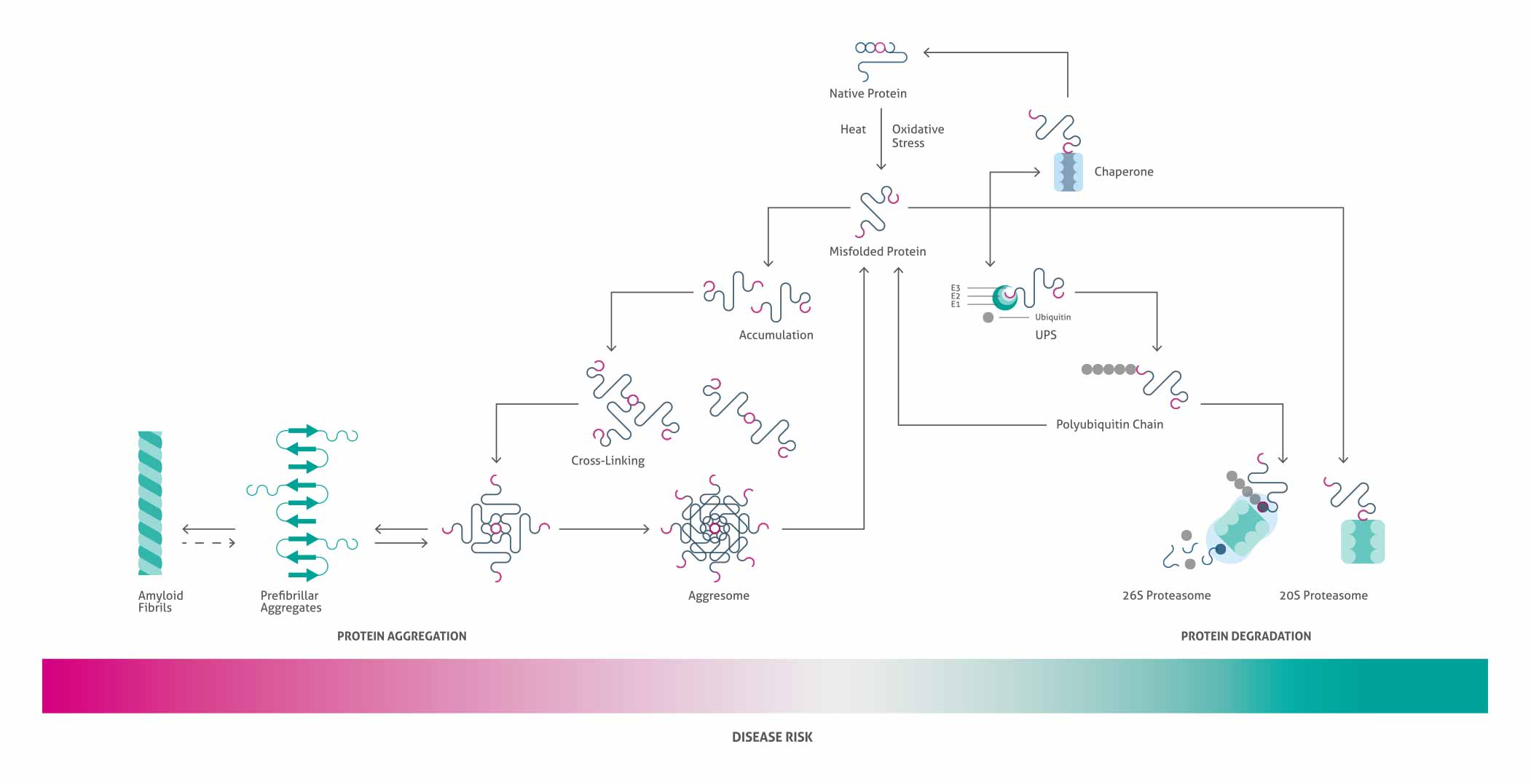
Protein misfolding events occur in healthy cells, but cellular mechanisms are in place that either help misfolded proteins refold correctly (e.g., protein chaperones) or remove them by protein degradation - see diagram above. This protein “triage” system is essential to maintaining the health of a cell. When this system malfunctions, or is overwhelmed by misfolded proteins, the balance shifts from protein refolding and/or degradation toward protein accumulation. Diseases characterized by fibrillar protein deposits show the extreme result of protein aggregation, where proteins in abnormal beta-sheet conformations cross-link irreversibly to form long, insoluble fibrillary complexes.
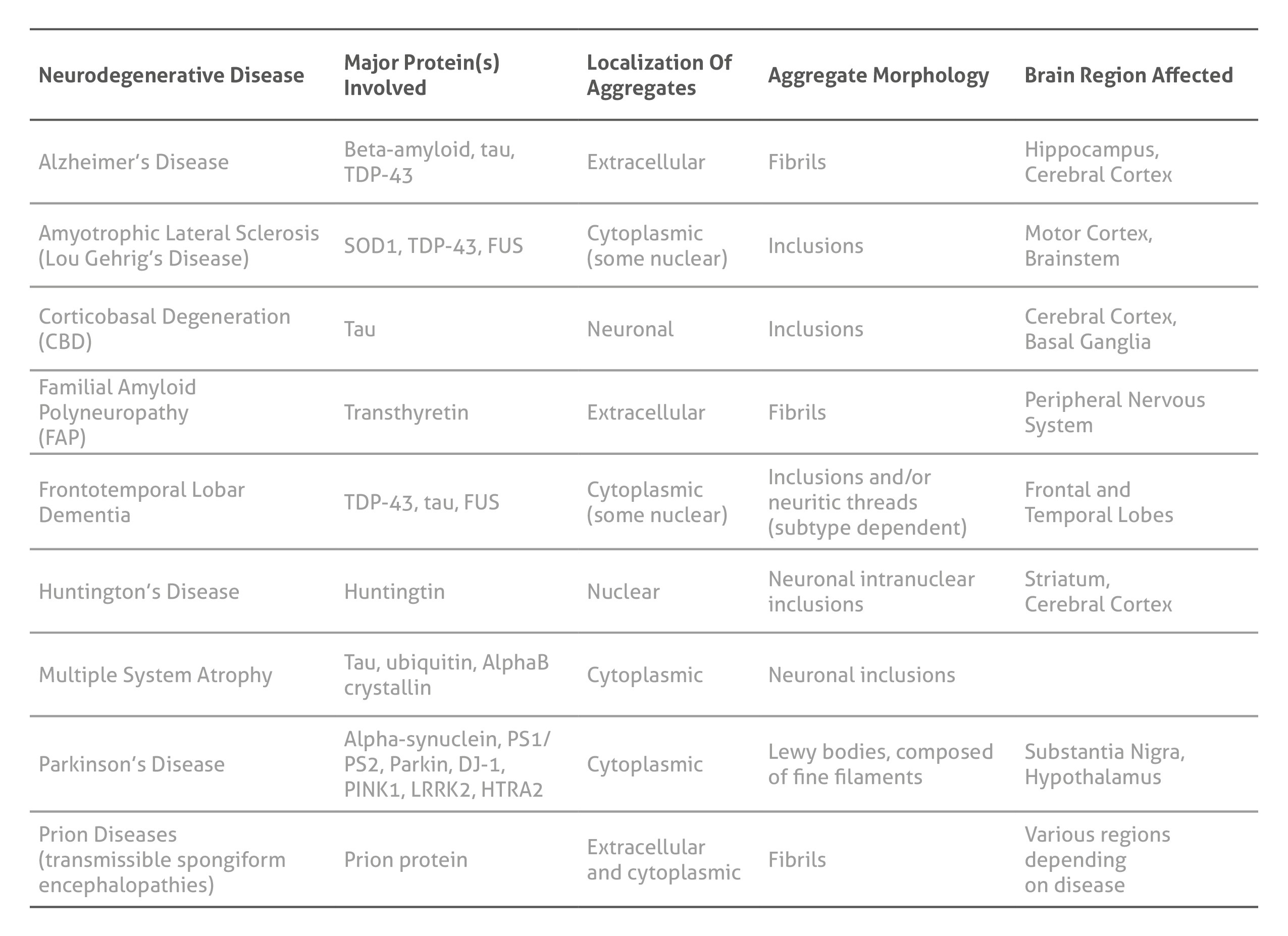
Neurodegenerative diseases and their pathologies
Download: Antibodies for Neuro-related Diseases full catalog |
Alzheimer's Disease
Alzheimer’s disease (AD) is a chronic disorder that slowly destroys neurons and causes serious cognitive disability. It is the most common form of dementia. AD is associated with senile plaques and neurofibrillary tangles (NFTs). Amyloid-beta, a major component of senile plaques, has various pathological effects on cell and organelle function. Extracellular amyloid-beta oligomers may activate caspases through the activation of cell surface death receptors. Alternatively, intracellular amyloid-beta may contribute to pathology by facilitating tau hyper-phosphorylation, disrupting mitochondria function, and triggering calcium dysfunction. To date, genetic studies have revealed four genes that may be linked to autosomal dominant or familial early-onset AD (FAD). These four genes include amyloid precursor protein (APP), presenilin 1 (PS1), and presenilin 2 (PS2). All mutations associated with APP and PS proteins can lead to an increase in the production of amyloid-beta peptides, specifically the more amyloidogenic form, amyloid-beta 42. FAD-linked PS1 mutation downregulates the unfolded protein response and leads to vulnerability to ER stress.
Download: Alzheimer's disease signaling pathway poster |
| TAU |
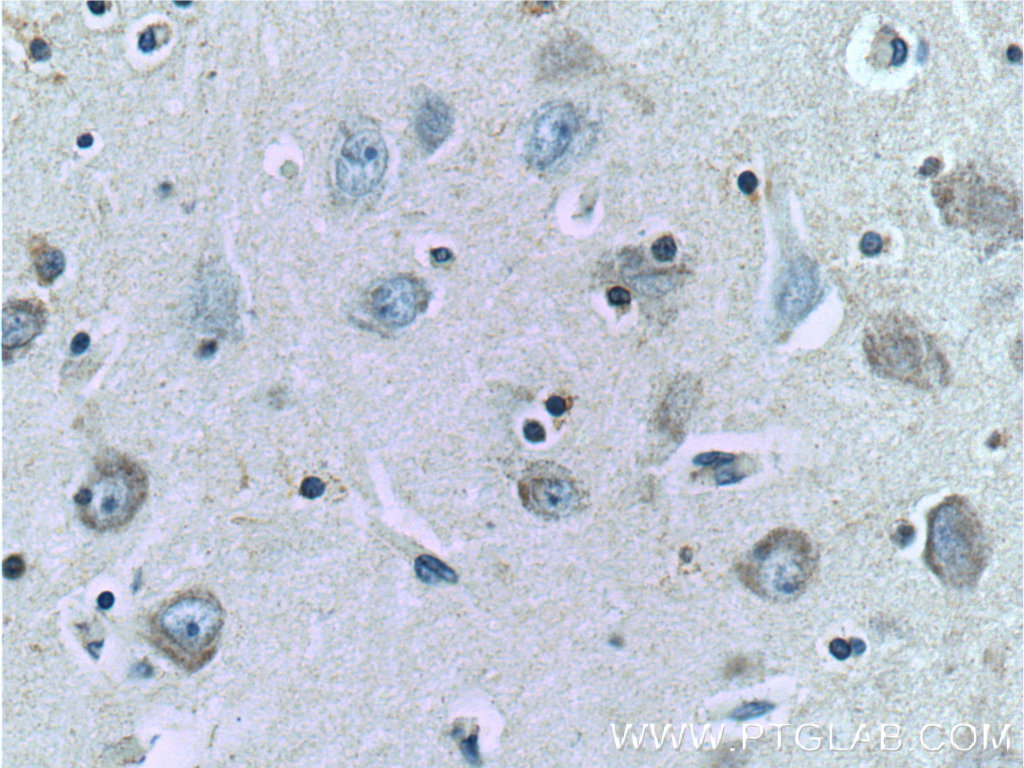 |
| IHC of paraffin-embedded human brain tissue slide using 10274-1-AP (TAU Antibody) at dilution of 1:200 (under 40x lens) |
| TDP-43 |
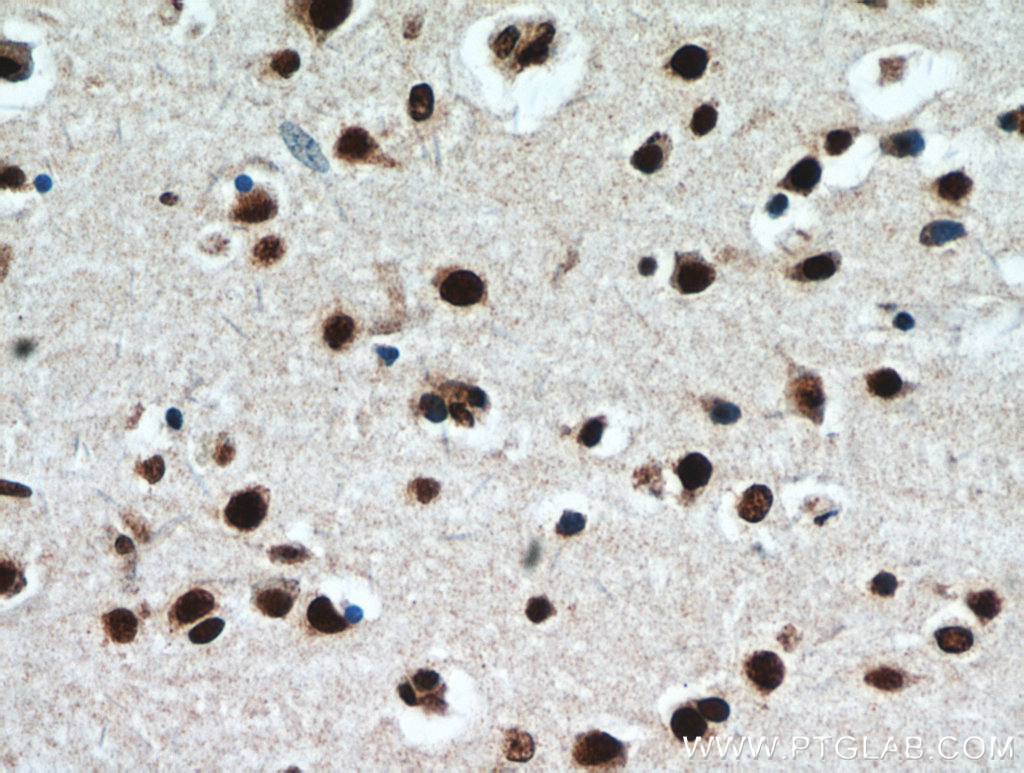 |
| IHC of paraffin-embedded human brain using 10782-2-AP (TARDBP antibody) at dilution of 1:50 (under 40x lens) |

Parkinson's Disease
Parkinson's disease is a long term disorder of the central nervous system which mainly affects the motor system of the CNS. Mutations in the a-synuclein gene are responsible for some familial forms of Parkinson's disease. The pathological hallmark of PD is an aggregation of synuclein in the form of Lewy bodies.
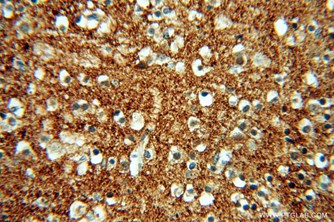 |
|
IHC of paraffin-embedded human brain using 10842-1-AP (SNCA antibody) at dilution of 1:50 (under 40x lens) |
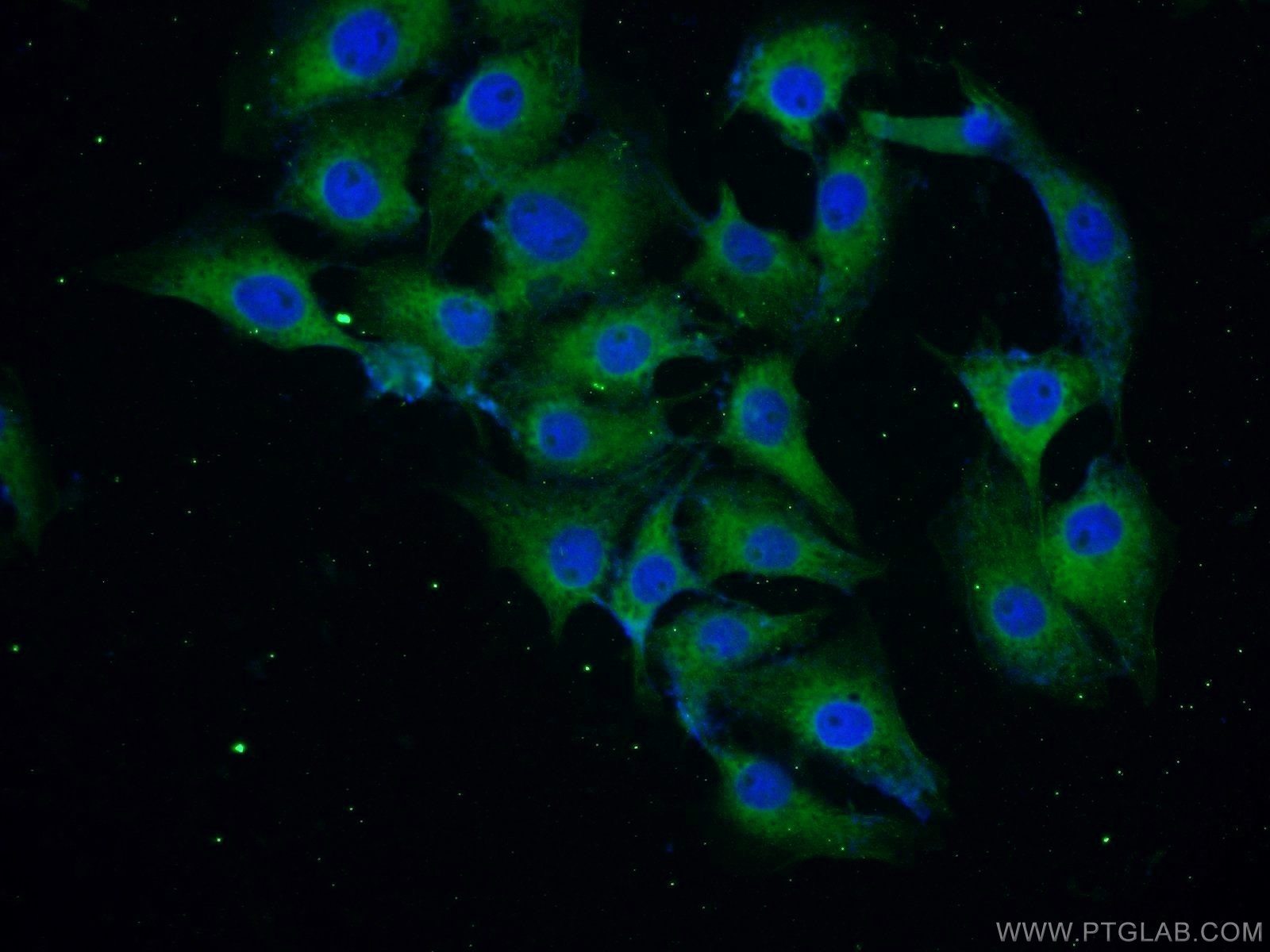 |
|
IHC analysis of SH-SY5Y cells using 10842-1-AP (a-Synuclein Antibody) at dilution of 1:25 and Alexa Fluor 488-conjugated AffiniPure Goat Anti-Rabbit IgG(H+L) |
Huntington's Disease
Huntington's disease is an autosomal dominant progressive neurodegenerative disorder characterized by progressive chorea, cognitive impairment and behavioural difficulties. Huntington's disease is caused by mutations in the HTT gene which provides the instructions for making the protein huntingtin.

积分商城

