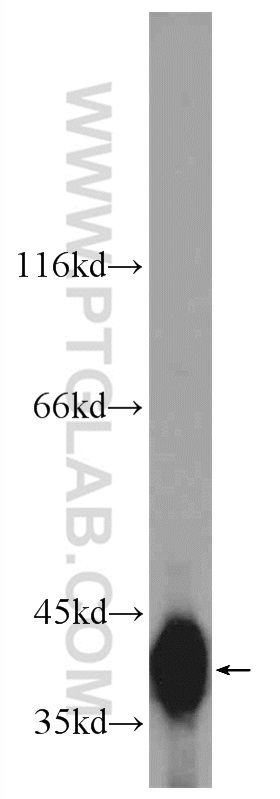
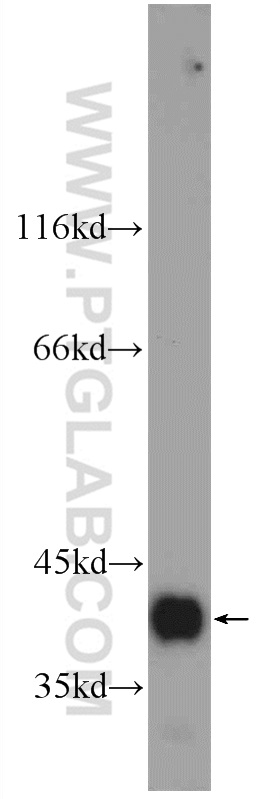
验证数据展示
 with sh-Control and sh-SMN transfected HEK-293 cells.")
 at dilution of 1:2000 incubated at room temperature for 1.5 hours.")
.")
.")
 at dilution of 1:50 and Alexa Fluor 488-conjugated Goat Anti-Rabbit IgG(H+L).")
经过测试的应用
| Positive WB detected in | K-562 cells, HepG2 cells, HEK-293 cells, Jurkat cells |
| Positive IHC detected in | human testis tissue Note: suggested antigen retrieval with TE buffer pH 9.0; (*) Alternatively, antigen retrieval may be performed with citrate buffer pH 6.0 |
| Positive IF/ICC detected in | HepG2 cells |
推荐稀释比
| 应用 | 推荐稀释比 |
|---|---|
| Western Blot (WB) | WB : 1:1000-1:4000 |
| Immunohistochemistry (IHC) | IHC : 1:50-1:500 |
| Immunofluorescence (IF)/ICC | IF/ICC : 1:20-1:200 |
| It is recommended that this reagent should be titrated in each testing system to obtain optimal results. | |
| Sample-dependent, Check data in validation data gallery. | |
发表文章中的应用
| WB | See 1 publications below |
| IF | See 1 publications below |
产品信息
22329-1-AP targets SMN in WB, IHC, IF/ICC, ELISA applications and shows reactivity with human samples.
| 经测试应用 | WB, IHC, IF/ICC, ELISA Application Description |
| 文献引用应用 | WB, IF |
| 经测试反应性 | human |
| 文献引用反应性 | human |
| 免疫原 | SMN fusion protein Ag17798 种属同源性预测 |
| 宿主/亚型 | Rabbit / IgG |
| 抗体类别 | Polyclonal |
| 产品类型 | Antibody |
| 全称 | survival of motor neuron 2, centromeric |
| 别名 | SMN1,SMN, SMN1, Gemin-1, Gemin 1, Component of gems 1 |
| 计算分子量 | 282 aa, 30 kDa |
| 观测分子量 | 38 kDa |
| GenBank蛋白编号 | BC000908 |
| 基因名称 | SMN |
| Gene ID (NCBI) | 6607 |
| RRID | AB_2879074 |
| 偶联类型 | Unconjugated |
| 形式 | Liquid |
| 纯化方式 | Antigen Affinity purified |
| UNIPROT ID | Q16637 |
| 储存缓冲液 | PBS with 0.02% sodium azide and 50% glycerol , pH 7.3 |
| 储存条件 | Store at -20°C. Stable for one year after shipment. Aliquoting is unnecessary for -20oC storage. |
背景介绍
Spinal muscular atrophy (SMA) is an autosomal recessive neurodegenerative disease characterized by loss of anterior horn cells in the spinal cord and concomitant symmetrical muscle weakness and atrophy (PMID: 16364894 ). SMA is caused by deletion or mutations of the survival motor neuron (SMN1) gene. SMA patients lack a functional SMN1 gene, but they possess an intact SMN2 gene, which though nearly identical to SMN1, is only partially functional (PMID: 17355180). A large majority of SMN2 transcripts lack exon 7, resulting in production of a truncated, less stable SMN protein (PMID: 10369862). The level of SMN protein correlates with phenotypic severity of SMA.
实验方案
| Product Specific Protocols | |
|---|---|
| WB protocol for SMN antibody 22329-1-AP | Download protocol |
| IHC protocol for SMN antibody 22329-1-AP | Download protocol |
| IF protocol for SMN antibody 22329-1-AP | Download protocol |
| Standard Protocols | |
|---|---|
| Click here to view our Standard Protocols |