
8 Top Tips for Subcellular Fractionation
8 technical tips for successful subcellular fractionation
By Alex Ryan
Subcellular fractionation is a method that dissects cells into their various organelles. High concentrations of sucrose are used to separate cell fractions based on their density. It is useful for looking at a single type of organelle in isolation, and allows processes to be studied in a cell free environment, without interference.
Here are my tips for successful subcellular fractionation...
1. Know your protein
Firstly, and most importantly, know your protein. This phrase is going to come up a lot, but don’t worry if you forget it: I will be repeating it. One of the first things this knowledge will help you to identify is whether or not fractionation is even necessary. There are plenty of reasons to carry out fractionation; you may want to look at protein translocation, membrane recruitment and sequestration, or as mentioned above investigate a single process without other cell functions interfering. If you just want to look at a protein in isolation, consider a method like immunoprecipitation instead.
From the outset, the first thing you need to know about your protein when planning your fractionation experiment is where you expect it to be. There are different types of fractionation (which we’ll get to later), and the type you choose will depend on where your protein is likely to be, and what you want to do with it. You also need to know the characteristics of your protein, as its function could also play a part in choosing your method. Transmembrane proteins can be very, shall we say, temperamental when it comes to purification. I know from personal experience that the insulin responsive glucose transporter, GLUT4, is a very grumpy protein.
2. Choose your method wisely
This comes from knowing your protein (see above). There are several methods of fractionation, leading to a different number of fractions. This all depends on knowing your protein, and what you wish to do with it… A fairly simple subcellular fractionation method can leave you with a crude membrane fraction [1]. Using this method you end up with three fractions: a nuclear fraction, a single membrane fraction and a very dilute cytoplasmic fraction. Cells are swollen with a hypotonic buffer, and the nuclei are spun out. Spinning the supernatant in a relatively low concentration of sucrose (0.34M) leaves a membrane pellet, and a cytoplasmic supernatant.
Removing the Triton X-100 from the homogenization buffer, as reported in Wali et al. [1], is an ideal method for looking at the activity of proteins at the membrane, especially if those proteins are recruited to membranes when activated (e.g. phospholipases C). Otherwise, this method provides perfect samples for Western blot analysis. Further centrifugation can be carried out on the cytoplasmic fraction to obtain individual organelles [2]
More thorough fractionation can be achieved using a greater concentration of sucrose (1.2M), and several separate centrifugations [3]. In this instance you can separate fractions into high density lipid fractions (i.e. rough endoplasmic reticulum) and low density lipid fractions (i.e. smooth endoplasmic reticulum, the Golgi and vesicles). Depending on the length of centrifugation, it is possible to isolate individual organelles [4]. These fractions are ideal for Western blotting if you are looking at trafficking throughout the cell.
3. Lysis buffer vs. swelling buffer
As you may have noticed above, hypotonic swelling buffers are key to subcellular fractionation of cell membranes. Lysis buffers generally contain a detergent, such as SDS, or an acid to break down the cells. Detergents play havoc with lipids in membranes, and cause the membranes to practically disappear. Whereas, swelling buffers are hypertonic solutions that cause the cell to gently “pop” through osmosis. If membrane integrity is important to your experiment then you should never use a lysis buffer.
4. Are you looking at protein activity?
This consideration is important if you are studying the activity of a protein. If so, it is essential that protein activity is not compromised at all. Many buffers contain phosphatase inhibitors, EDTA or EGTA, and detergents, all of which can prevent proteins from working properly. If you are using Western blots to analyze your fractions it doesn’t matter if the proteins are non-functional and denatured, but if you intend to investigate a cellular process biochemically then it is imperative that all proteins are able to function accurately…so put down the additives.
5. Know your fraction
If you are carrying out Western blotting on individual fractions, or organelles, it is important that you can confirm you are looking at the correct organelle fraction. Luckily antibody companies like Proteintech have many organelle- or fraction-specific control antibodies (see below), which can be used to verify that the isolated cell component you’re looking at is the right one. Some fractions are tricky to confirm (such as plasma membrane fractions) in these instances consider using a panel of both positive and negative markers to assess purity.
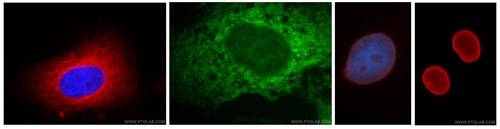
Antibodies are useful for the validation of fractions via Western blot. This image shows various organelles visualised by immunofluorescence imaging using some of Proteintech's antibodies, which are also suitable for use as fraction loading probes on Western blot membranes. L-R Hela cells stained with anti-alpha tubulin (red), the ER of A431 cells is stained with anti-calnexin (green), and anti-lamin A/C (3rd from left) and anti-lamin B1 (far right) stain the nuclear envelope (red) of HepG2 cell samples.
6. Igepal CA-630 is your friend!
Igepal CA-630 can protect transmembrane proteins from degrading. If you are having difficulty seeing your transmembrane protein on a Western blot, then adding 0.1% (v/v) Igepal CA-630 may help you.
7. Plan ahead
Subcellular fractionation can take a long time... In the GLUT4 example I mention above, the experiment actually became a three person job! My Supervisor would stimulate the sample when she arrived at about 7:30am. I would come in at 8:30, carry out the fractionation, protein assays, and prepare samples for Western blotting. I’d then leave another PhD student with the samples “boiling” at 60˚C for 30 minutes at about 5pm. He would then immediately carry out the gel electrophoresis, and transfer. Meaning the entire experiment would take roughly 14 hours, and this excludes the subsequent analysis by Western blotting!
It is also important that all fractions are kept on ice as much as possible, and all buffers are ice‑cold. Plus, ultracentrifuges can take a long time to cool down, so it is important to get an early start. Better reacquaint yourself with your alarm clock and be nice to your colleagues!
8. Know your protein
Yep, I told you I’d be repeating it...It’s important.
Related antibodies
| Organelle | Catalog No.(s) | Target |
| Cytoskeleton | 66031-1-AP | α tubulin |
| Cytoskeleton | HRP-66031 (HRP-conjugated) | α tubulin |
| Cytoskeleton | 60008-1-Ig/20536-1-AP | β actin |
| Cytoskeleton | HRP-60008 (HRP-conjugated) | β actin |
| Endoplasmic reticulum | 10427-2-AP | Calnexin |
| Endoplasmic reticulum | 11587-1-AP | GRP78/HSP70 |
| Golgi | 11308-1-AP | GM130 |
| Golgi | 12255-1-AP/66170-1-Ig | RCAS1/EBAG9 |
| Membranes | 20648-1-AP | E-Cadherin |
| Membranes | 14418-1-AP | Na+/K+ ATPase |
| Membranes | 17435-1-AP/66171-1-Ig | Transferin |
| Mitochondrion | 11242-1-AP | COX IV 1 |
| Mitochondrion | 10866-1-AP | VDAC1/Porin |
| Nuclear envelope | 10298-1-AP | lamin A/C |
| Nuclear envelope | 12987-1-AP | Lamin B1 |
| Nucleus | 20813-1-AP | LSD1/KDM1 |
| Nucleus | 11231-1-AP | SETDB1/ESET |
References:
1. Wali, R. K., Baum, C. L., Sitrin, M. D. and Brasitus, T. A. (1990) 1,25(OH)2 vitamin D3 stimulates membrane phosphoinositide turnover, activates protein kinase C, and increases cytosolic calcium in rat colonic epithelium. J. Clin. Invest. 85, 1296–303.
2. Schröter, C. J., Braun, M., Englert, J., Beck, H., Schmid, H. and Kalbacher, H. (1999) A rapid method to separate endosomes from lysosomal contents using differential centrifugation and hypotonic lysis of lysosomes. J. Immunol. Methods 227, 161–168.
3. Grainger, D. L., Tavelis, C., Ryan, A. J. and Hinchliffe, K. A. (2011) Involvement of phosphatidylinositol 5-phosphate in insulin-stimulated glucose uptake in the L6 myotube model of skeletal muscle. Pflugers Arch. 462, 723–32.
4. Graham, J. M. (2001) Isolation of Golgi membranes from tissues and cells by differential and density gradient centrifugation. Curr. Protoc. Cell Biol. Chapter 3, Unit 3.9.






