
Rett syndrome - the second leading cause of mental retardation in women
In 1966, Rett syndrome was for the first time described as a clinical issue.
A few words about Rett syndrome
Rett syndrome (RTT) is a cause of mental retardation affecting 1 in 10,000 female births, making it the second leading cause of mental retardation in girls. In 1999 Zoghbi lab discovered the genetic basis of Rett syndrome. In 95% of cases, a mutation of MeCP2 is the cause of classical RTT (1).
MeCP2 is a nuclear protein that is expressed widely in different tissues but is most abundant in the neurons of a mature nervous system (1, Figure 1).
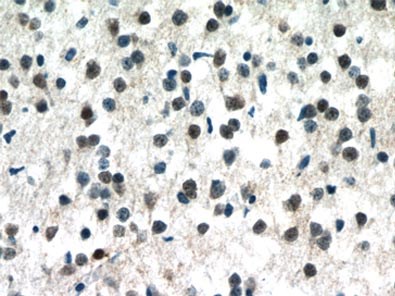
Figure 1. Immunohistochemistry of paraffin-embedded human brain tissue slide using 10861-1-AP (MECP2 Antibody) at a dilution of 1:50 (under 40x lens).
Although the function of MeCP2 remains unknown, it is considered likely to regulate gene expression, either through the silencing or activation of specific genes or by more global regulation of transcriptional processes (2). RTT was the first neurodevelopmental disorder related to epigenetics. The CDKL5 and FOXG1 genes have since been associated with variant forms of Rett syndrome (3).
Clinical features of Rett syndrome
RTT is a postnatal progressive disease with a normal prenatal and perinatal period. Rett patients also have proper brain development during the first 6 to 18 months of age, which is known as the stagnation stage. During this time girls achieve normal neurodevelopment, motor function, and communication skills. However, developmental regression subsequently appears (4, 5).
During the active regression stage of the disease, which occurs between the ages of 1 and 4 years, girls lose the ability to speak and the purposeful use of their hands. Very often they also show autistic features, related mostly to a reduction in interpersonal contact. There is a deceleration in the rate of head growth caused by microcephaly. Neurological evaluations show cortical hyperexcitability on an electroencephalogram, indicating a loss of developmental features.
In the third, pseudostationary, phase of the disease, which occurs between the ages of 4 and 7 years, girls become more alert but with more abnormalities, including teeth grinding, screaming fits, severe scoliosis, reduced somatic growth, low mood, or night crying and laughing. Epilepsy is also briefly evident at this time. Stereotyped hand movements are seen, typically in the form of hand-wringing and washing but also hand tapping/clapping or clasping. Eye contact, while initially reduced, somehow returns, and a child may come to look more alert and joyful, with typical eye-pointing behavior to express needs and wishes.
During the late motor deterioration stage, occurring at ages 5 to 15 years and older, girls begin to have cardiac abnormalities (bradycardia and tachycardia) that carry a high risk of sudden death due to respiratory dysfunction. In many cases, lack of mobility leads to a state called frozen rigidity. However, some girls never lose the ability to walk and remain at stage 3 throughout their lives (summary in Figure 2).
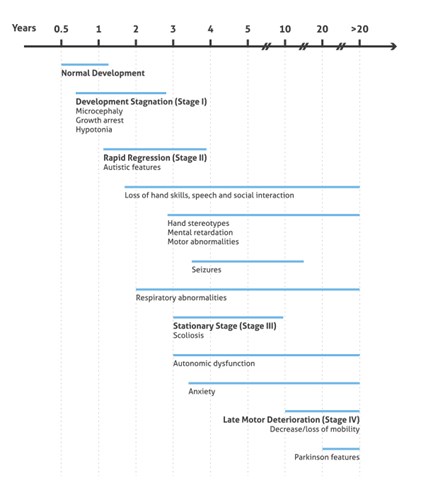
Figure 2. Illustration presenting the onset and progression of RTT Clinical Phenotypes (based on 3).
Abnormalities and neuropathology in Rett syndrome patients
The weight of the brain of a patient affected by RTT is significantly lower than that of the brain of age-matched healthy controls; however, the weight does not decrease significantly with age (6). The neurons are smaller and more densely packed, resulting in a reduced brain volume.
The main changes relate to the prefrontal, posterior temporal, and posterior occipital regions. Neuropathology has also been observed in cerebrospinal fluid, including acetylcholine (7, 8), dopamine (9, Figure 3), serotonin (10), glutamate (11), or nerve growth factor (12).
Also, levels of microtubule-associated protein 2 (MAP2, Figure 4) have been found to be reduced by the loss and gain of MeCP2 expression in accordance with the involvement of MeCP2 in shaping dendritic morphology.
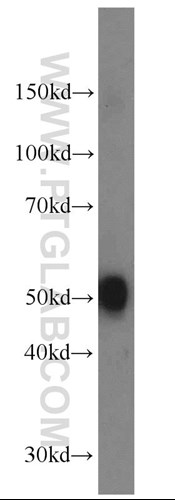
Figure 3. Mouse brain tissue was subjected to SDS PAGE followed by western blot with 10166-1-AP (DOPA decarboxylase antibody) at a dilution of 1:1000 incubated at room temperature for 1.5 hours.
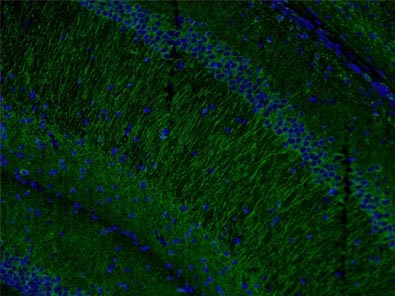
Figure 4. Immunofluorescent analysis of (4% PFA) fixed mouse brain tissue using 17490-1-AP (MAP2 antibody) at a dilution of 1:50 and Alexa Fluor 488-conjugated AffiniPure Goat Anti-Rabbit IgG(H+L).
Animal models of Rett syndrome
Mice models are helping us to understand the mechanisms and pathology of many diseases due to mouse brain having a similar structure to a human brain.
There is still no effective treatment available for Rett syndrome. Mecp2 knockout mice have a range of physiological and neurological abnormalities that resemble the human syndrome and can be used as a model to interrogate new clinical therapies targeting Rett abnormalities.
Written by Karolina Szczesna; PhD in Biomedicine
Senior Product Manager, Proteintech Ltd.
References:
- Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2.
- MeCP2, a key contributor to neurological disease, activates and represses transcription.
- The story of Rett syndrome: from clinic to neurobiology.
- Clinical manifestations and stages of Rett syndrome.
- The clinical recognition and differential diagnosis of Rett syndrome.
- Neuropathology of Rett syndrome.
- Rett syndrome: neurobiological changes underlying specific symptoms.
- Choline acetyltransferase activity and vesamicol binding in Rett syndrome and in rats with nucleus basalis lesions.
- Reduction of biogenic amine levels in the Rett syndrome.
- Polysomnography in the Rett syndrome.
- High levels of cerebrospinal fluid glutamate in Rett syndrome.
- Low levels of nerve growth factor in cerebrospinal fluid of children with Rett syndrome.





